
Urgent Implementation of UDI Comprehensive Interpretation Provided in a Timely Manner
The National Medical Products Administration (NMPA) issued an announcement on September 17, 2021, titled "Announcement on the Implementation of the Second Batch of Medical Device Unique Identification Work," which explicitly includes all Class III medical devices (including in vitro diagnostic reagents) in the second batch of unique identification implementation and sets a deadline of June 1, 2022.
At the national level, all Class III medical devices in the country must complete UDI implementation by June 1 of this year. This also signifies that the implementation for Class I and Class II medical devices nationwide is one step closer.
Medical devices encompass a wide range of instruments or equipment used for treatment or diagnosis, directly in contact with patients. From simple contact lenses to more complex devices like pacemakers, the frequency of using medical devices in daily life is very high.
Class III Medical Devices - High Risk
Includes implantable pacemakers, pulse generators, HIV diagnostic tests, automated external defibrillators, endogenous implants, and long-term wear contact lenses.
Class II Medical Devices - Medium Risk
Includes acupuncture needles, daily wear contact lenses, electric wheelchairs, infusion pumps, surgical drapes, and implantable radiofrequency transponder systems for patient identification and health information.
Class I Medical Devices - Low Risk
Includes elastic bandages, examination gloves, and handheld surgical instruments.
Medical device enterprises plan their compliance processes based on the classification of their products. The intensity of UDI regulatory control varies by category, with higher categories subject to stronger controls and earlier compliance deadlines. The completion deadline for Class III medical devices is earlier than for the other two classes.
"The Unique Device Identification (UDI) refers to a code composed of numbers, letters, or symbols attached to a medical device product or its packaging, used for the unique identification of the medical device." (Article 3 of the "Medical Device Unique Identification System Rules") "The Unique Device Identification includes the device identifier (DI) and the production identifier (PI)." (Article 7 of the "Medical Device Unique Identification System Rules")
The Unique Device Identification System (UDI System) is currently a hot topic in the field of medical device regulation. The United States and the European Commission issued UDI system-related regulations in 2013 and 2017, respectively. China also released the "Medical Device Unique Identification System Rules" in August 2019, aiming to establish a comprehensive identification system for the entire lifecycle of medical devices.
UDI Identification Specifications under the GS1 Standard
Based on the risk level and traceability requirements of medical devices, the UDI code structure consists of the DI (Device Identifier - static information) and PI (Production Identifier - dynamic information). The following is an example of the UDI structure based on the GS1 standard:
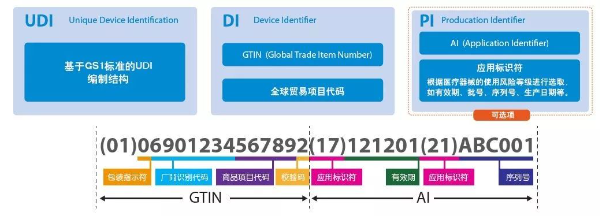
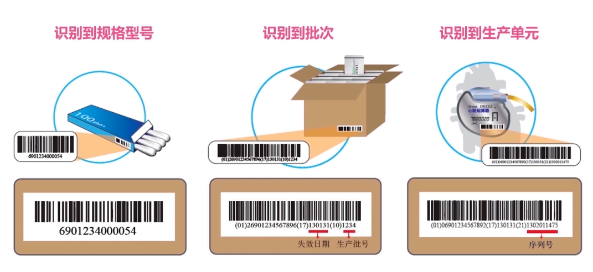
Due to differences in the risk levels and regulatory traceability requirements of medical devices, the UDI structure varies accordingly. The UDI can be represented by the DI alone or by a combination of DI and PI, as shown below:
According to the medical device unique identification database, as of February 15, 2022, there are 1,074,741 DIs using the GS1 standard in China, accounting for 91% of the total.
The application of UDI can improve the efficiency of recall procedures, reduce medical accidents, and increase inventory visibility and supply chain security.
Applying UDI to each product during the manufacturing phase provides critical information for supply chain management systems, such as what the product is, where and when it was produced, its current location, and its circulation path. In the event of a product failure requiring a recall, UDI is crucial for the regulatory chain to trace the source.

